
2.7.ANTIBACTERIENS SYNTHETIQUES
- LES SULFAMIDES (OU SULFONAMIDES)
L’intérêt des antibactériens sulfamides nait en 1933 lorsqu’une équipe de l’institut Pasteur démontre l’activité de la p-aminobenzènesulfonamide ou sulfanilamide, nom contracté en
« sulfamide ». Mais la découverte d’autres antibiotiques plus efficaces et moins dangereux fait reculer l’intérêt pour ces molécules. Actuellement les sulfamides utilisables par voie générale sont souvent associés à d’autres molécules (trimétoprime dans le cotrimoxazole par exemple).
- Mode d’action: inhibition de la synthèse de l’acide folique, nécessaire à la synthèse des purines. Les sulfamides profitent de leur ressemblance au PABA. En La membrane des bactéries étant imperméable à l’acide folique, elles ne pourront pas l’importer de l’extérieur. La carence en acide folique entraina logiquement un déficit en purines et donc en acides nucléiques. Les cellules humaines sont très peu affectées par l’action des sulfamides car elles sont perméables à l’acide folique issu de l’alimentation.
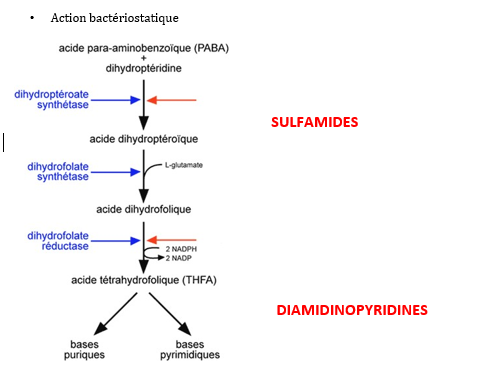
La ressemblenace structurelle des sulfamides avec l’acide paraaminobenzoïque (PABA) est à la base de leur action inhibitrice. En présence de grande quantités de sulfamide, la bactérie les utilise de préférence au PABA mais cela n’aboutit pas à la synthèse de l’acide folique.

Les résistances se développent par
- hyperproduction d’acide para-aminobenzoïque suite à une mutation chromosomique. La loi d’action de masse jouant, la bactérie redemarre la synthèse de l’acide folique en augmentant la concentration en PABA.
- modification de la structure de la dihydrofolate synthétase suite à une mutation chromosomique
- production de dihydroptéroate synthétase à affinité réduite suite à l’acquisition d’un plasmide
- réduction de la perméabilité bactérienne aux sulfamides suite à l’acquisition d’un plasmide
- modification de la structure de la dihydrofolate synthétase suite à une mutation chromosomique
Pharmacocinétique
Bonne résorption digestive
Diffusion dans les liquides interstitiels et le LCR ; Cependant cette propriété n’est plus très utile en raison des résistances dévelopées par les germes responsables des méningites (sauf Listeria monocytogenes)
Diffusion dans tous les tissus y compris le LCR et l’œil
Élimination essentiellement rénale, ce qui justifie leur utilisation dans les infections urinaires.
Types
- Sulfisoxazole: absorption et élimination rapides
- Associé à l’erythromycine (pediazole) pour le traitement des otites moyennes aigues chez les enfants
- Sulfamethoxazole: proche de sulfisoxazole; associé à la trimetoprime (cotrimoxazole)
- Sulfadiazine: usage générale et en topique en prévention des infections des brûlures (sufadiazine argentique: flammazine®)
- Sulfadoxine: tong t½, asocié à la pyriméthamine (fansidar®) pour le traitement du paludisme
Effets indésirables:
- troubles hématologiques: anémie mégaloblastique, hémolyse en cas de déficience en G6-PD
- Allergies
Indications:
- Toxoplasmose: sulfadiazine (4x1g/jPO) +pyriméthamine (75mg charge puis 25mg/jPO) + acide folinique!!!
- Nocardiose: sulfadiazine ou sulfisoxazole (6-8g/j) besoin de continuer plusieurs mois après le fin des symptomes)
Co-trimoxazole (trimetoprime+ sufaméthoxazole)
- La Synergie entre les deux est optimale pour 20 parts de sulfamethoxasole contre 5 parts de trimetoprime. C’est ainsi que l’on aura des présentations en 480mg (400/80), 960mg (800/160).
- Résistances en croissance à cause de l’utilisation massive et mauvaise. Il y a des craintes que les personnes VIH+ recevant le cotrimoxazole au long cours en prévention des IO soient source de germes résistants. Des études en Uganda et dans d’autres pays semblent montrer qu’il n’y a pas à s’inquiéer sur ce sujet. Il n’y a pas augmentation des résistances. La prophylaxie au cotrimoxazole est efficace même dans les zones de forte résistance à ce produit.
- PK: bonne absorption. En administrant 800/160mg PO on obtient au bout d’une heure la même concentration plasmatique qu’après administration de la même dose en IV.
- Résistances en croissance à cause de l’utilisation massive et mauvaise. Il y a des craintes que les personnes VIH+ recevant le cotrimoxazole au long cours en prévention des IO soient source de germes résistants. Des études en Uganda et dans d’autres pays semblent montrer qu’il n’y a pas à s’inquiéer sur ce sujet. Il n’y a pas augmentation des résistances. La prophylaxie au cotrimoxazole est efficace même dans les zones de forte résistance à ce produit.
• Indications:
- Infections urinaires, gastro intestinaux
- Peumonie à Pneumocystis jiroveci chez les PVV:
- Curatif: fortes doses (T 15mg/Kg/J + S 100mg/Kg/J en 3 ou 4 Prises)
- Préventif: 960mg/j ou 3xpar semaine
- Prévention des infections chez les neutropéniques: 2 X 960mg.J
• Effets indésirables:
- réactions cutanées, troubles hématologiques, troubles digestifs, ictère,
- Plus fréquents chez les PVV. Assurer la désensibilisation puis continuer traitement.
Désensibilisation au cotrimoxazole
En cas d’allergie au cotrimoxazole et lorsque son administration est indispensable, il est nécessaire de procéder à la désensibilisation :
- Préalables: la désensibilisation ne peut se faire qu’en salles des soins intensifs ; un abord veineux est placé et le nécessaire pour la prise en charge du choc anaphylactique est mis à portée de main. L’on procédera comme suit:
- 4mg de sulfamethoxazole toutes les 6 heures
- Doubler la dose tous les 24 heures jusqu’à atteindre la dose thérapeutique désirée
Après la désensibilisation le patient peut encore présenter des réactions allergiques mais elles seront plus bénignes et plus rares ; il peut aussi arriver qu’il ne présente plus de réaction allergique.
- 2.7.1. LES QUINOLONES
Les quinolones sont actives sur une grande variété d’infections. Ils agissent par inhibition de la topoisomérase IV ou l’ADN gyrase.
L’ADN gyrase : les deux brins de la double hélice de l’ADN doivent se séparer pour permettre sa réplication (nécessaire avant la division cellulaire) ou sa transcription (pour la synthèse des protéines). Mais cette séparation entraine une sur-spiralisation ou super-enroulement positif qui crée un obstacle mécanique. Les bactéries sont équipées de l’enzyme dénommée ADN-gyrase dont le rôle est d’empêcher cela en créant des super-spirales négatives et permettre ainsi que la division cellulaire ou la production des protéines ait lieu. En inhibant l’ADN gyrase, les quinolones empêchent ainsi la synthèse des protéines bactériennes. Pour les cellules des eucaryotes, c’est la topoisomérase II qui joue le rôle de l’ADN gyrase.
La topoisomérase IV : elle a pour rôle de séparer les chaines d’ADN résultant de la réplication.
L’ADN gyrase est la cible des quinolones pour la plupart des bactéries Gram- tandis que la topoisomérase IV l’est pour la plupart des bactéries Gram+.
Spectre : staphylocoques (mais pas les Méti-R, streptocoques (uniquement mixifloxacine, levofloxacine et gatifloxacine), bactéries intracellulaires : chalamydia, mycoplasma, legionella, mycobactérium (dont tuberculosis).
Les résistances se développent par mutation sur les gènes codant pour la topoisomérase IV ou l’ADN gyrase ou encore par transport actif hors de la bactérie.
Pharmacocinétique
Bonne absorption pour la majorité des quinolones, la biodisponibilité dépasse 50% ; parfois 95%. Le volume de distribution est souvent élevé avec concentration dans les urines, la bile, la prostate et les poumons et même dans les macrophages les neutrophiles. L’excrétion se fait souvent par les reins. La ciprofloxacine, l’ofloxacine et la péfloxacine passent dans le lait maternel.
Indications
- Infections urinaires : les quinolones de 1ère génération sont considérées comme des antiseptiques urinaires. Les autres quinolones sont aussi efficaces dans les infections urinaires.
- Prostatite aigue et chronique (4-6 semaines de traitement dans la prostatite chronique) (norfloxacine, ciprofloxacine et ofloxacine)
- Infections gastro-intestinales et abdominales : entérites bactériennes, (shigellose, salmonelloses…), péritonites (souvent en association avec les nitroimidazolés)
- Infections respiratoires : cependant les quinolones de 1er et 2ème générations sont inactives contre le S. pneumoniae et les anaérobies ; mais les nouvelles quinolones notamment gatifloxacine et moxifloxacine sont actives contre ces germes.
- Infections des os, des articulations et des tissus mous
- Autres indications : anthrax, tuberculose multi résistant, mycobactéries atypiques
- 1ère génération (désinfectants urinaires): acide nalidixique (negram), acide pipémidique (pipram)
- 2ème génération (fluoroquinolones, systémiques): norfloxacine, ciprofloxacine, ofloxacine
- 3ème génération (fluoroquinolones actifs sur les G-): sparfloxacine
Effets indésirables:
- Troubles digestifs, tendinite,
- Photosensibilité
- Allongement de QT rare
- Hypo ou hyperglycémies constatés chez les diabétiques avec la gatifloxacine (contre- indication)
- arthropathie chez les enfants. Il s’agit plus d’une crainte que de faits. L’effet indésirable a été constaté chez les animaux, mais il n’y a pas encore de confirmation de la survenue du même problème chez les enfants humains. Cependant, il serait prudent de n’utiliser les quinolones qu’en dernier recours chez les enfants.